
-
|
-
Multiplex Western blotting (WB) enables the simultaneous detection of several proteins on a single membrane. By probing the blot with differentially‑labeled fluorescent secondary antibodies, the expression levels of each target can be visualized in one experiment. This approach is especially valuable when quantitative analysis is required, because the intensity of every protein of interest is measured alongside an internal loading control (e.g., β‑actin, GAPDH, or total‑protein stain) under identical electrophoretic and transfer conditions. Consequently, variation arising from gel loading, transfer efficiency, or membrane handling is intrinsically corrected, yielding more reliable and reproducible quantification. Multiplex WB also conserves samples, reagents, and instrument time, making it the method of choice for high‑throughput or limited‑material studies.
In indirect fluorescent multiplex Western blots, primary antibodies should ideally come from different host species to prevent cross-reactivity of secondary systems. However
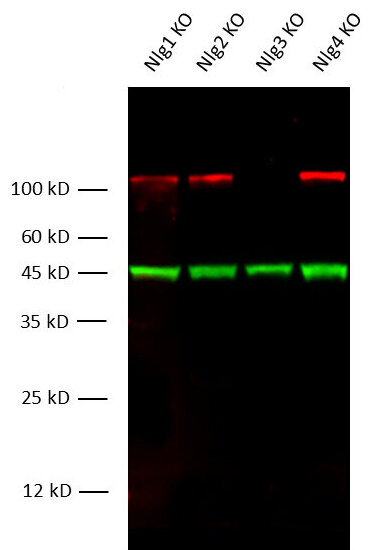
Figure 1: Fluorescent multiplex Western blot detection of Neuroligin3 in mouse brain lysates from different Neuroligin K.O. strains with rabbit anti-Neuroligin3 antibody (cat. no. 129 308, dilution 1:1000) and mouse anti-beta-Actin antibody (cat. no. 251 011, dilution 1:10000) as a loading control.
Immunoreactivity was revealed by fluorescent detection.
Figure 1: Fluorescent multiplex Western blot detection of Neuroligin3 in mouse brain lysates from different Neuroligin K.O. strains with rabbit anti-Neuroligin3 antibody (cat. no. 129 308, dilution 1:1000) and mouse anti-beta-Actin antibody (cat. no. 251 011, dilution 1:10000) as a loading control.
Immunoreactivity was revealed by fluorescent detection.
Western blot stripping is a post‑detection step that removes bound antibodies (both primary and secondary) from a nitrocellulose or PVDF membrane while leaving the immobilized target proteins intact. The cleared membrane can then be re‑blocked and probed again with a different set of antibodies, allowing multiple proteins to be visualized from a single electrophoretic run.
|
Buffer |
Formulation |
pH |
Conditions |
Pros |
Cons |
|
Low-pH glycine-SDS |
0.1M glycine-HCl |
2.2 |
30 min–1 h at room temperature (RT) |
very fast, works well for most HRP‑ or fluorescence‑conjugated antibodies; preserves membrane integrity; inexpensive |
high‑affinity antibodies may not be efficiently stripped; |
|
β‑mercaptoethanol / SDS (Harvey & McCulloch buffer) |
62.5 mM Tris‑HCl, 2 % (w/v) SDS, 0.5 M β‑ME, 0.01 % (v/v) Bromophenol blue |
6.8 |
65 °C water bath, 15–30 min |
strong stripping power - removes virtually any IgG; good for re‑probing |
harsh - can damage the membrane, especially nitrocellulose; β‑ME is toxic and odorous; may also partially elute transferred proteins after repeated cycles |
|
RIPA‑based stripping buffer |
150 mM NaCl, 1 % (v/v) NP‑40, 0.5 % (w/v) sodium deoxycholate, 0.1 % (w/v) SDS, 50 mM Tris‑HCl pH 7.5, 0.1 % (v/v) β‑ME |
7.5 |
55 °C, 20–30 min |
good compromise – strong enough for high‑affinity antibodies but milder than SDS‑β-ME; works for both chemiluminescent and fluorescent detection |
β‑ME is toxic and odorous |
|
Commercial “mild” stripping buffer (e.g., Thermo Scientific # 21059, LI‑COR REVERT 700) |
proprietary – typically contains a low‑pH glycine, mild non‑ionic detergent, and a reducing agent (often DTT or TCEP) |
2.5-3.0 |
RT, 5–15 min |
optimized for fluorescent Westerns (e.g., IR‑Dye antibodies); very low background after re‑probing; no β‑ME, no harsh SDS |
expensive |
Several practical points affect the success of successive probing.
PVDF membranes are generally more tolerant to high‑pH, high‑SDS, and reducing conditions than nitrocellulose, so they are preferred when multiple stripping cycles are anticipated.
Some epitopes are sensitive to alkaline or detergent exposure; if a band disappears after one round, a milder acidic stripping buffer (pH 2.2) or a lower SDS concentration should be tried. After each stripping step it is advisable to include a control lane that receives no new primary antibody; any residual band indicates incomplete removal of the previous antibodies.
Because the cumulative loss of protein can become noticeable after two or three cycles, the most abundant loading control (e.g., β‑actin, GAPDH, or a total‑protein stain) is often applied in the final round.
Certificates
ISO 9001 2015 Quality Management System and Green Lab Platinum certification level for sustaining laboratory processes.


Newsletter
Sign up for our newsletter and get the latest updates and news.